


Next: 7 Examples, examples_qe, inputs,
Up: User's Guide for the
Previous: 5 Restarting an interrupted
Contents
The directory tools contains a few tools that can be useful to build
structures of solids, surfaces, ribbons, and nanowires. Moreover it
contains some miscellaneous codes that give additional information
on the internal conventions of thermo_pw or further process its output.
Currently it contains the following programs:
- bravais_lattices.x tests the
module lattices.f90 of the library. Pre- sently it can read three
primitive lattice vectors of a Bravais lattice and find the ibrav
code and the celldm parameters of the input lattice. It
can also read two sets of primitive vectors and decide if they describe
the same Bravais lattice. In the positive case it gives the orientation
of one lattice with respect to the other.
For an example of its use see tools_input/bravais_lattice.in.
- crystal_point_group.x is a crystal point group calculator.
It can give several information about the crystallographic point groups,
such as the list of symmetry operations, the product of two rotation
matrices, the product table, the class structure, the character tables of
the irreducible representations of the point group and of the double
point group and the projective representations of the point group. It
gives the list of subgroups and supergroups of a given group and
the compatibility tables of a given group with its subgroups. It can
also decompose the Kronecker product representations. Finally it
can list the conjugate groups and calculate the intersection of two
point groups.
For an example of its use see tools_input/crystal_point_group.in.
- debye.x reads from input the Debye temperature and the
number of atoms per unit cell and writes in a file the thermodynamic
quantities (vibrational energy, free energy, entropy, and heat capacity) as
a function of temperature computed using the Debye model. When the
system has only one atomic type it writes the atomic B
factor
as a function of temperature computed using the Debye model.
For an example of its use see tools_input/debye.in.
- elastic.x reads the elastic constants of a
solid and computes the elastic compliances, the bulk modulus, and a few
poly-crystalline averages. It uses the thermo_pw library so the output is the
same.
For an example of its use see tools_input/elastic.in.
- epsilon_tpw.x generalizes the routine
epsilon.f90 of the QUANTUM ESPRESSO distribution. It calculates the
complex dielectric
constant of a solid as a function of the frequency for independent electrons
using the LDA or GGA eigenvalues. It is limited to insulators, but supports
norm-conserving, ultrasoft, and PAW pseudopotentials. It supports both scalar
relativistic and fully relativistic pseudopotentials and it uses the point
group symmetry of the solid to reduce the number of k-points.
For an example of its use see example14.
- gener_nanowire.x reads a two dimensional (2D)
Bravais lattice index and atomic coordinates an generates a sheet of type
(m, n)
. A sheet contained between the two vectors
C= m
a1 + n
a2
and T = p
a1 + q
a2
can be also generated and wrapped about C in a nanotube form
(a1
and a2
are the primitive lattices of the 2D Bravais
lattice).
For lattices that allow it, p
and q
can be determined automatically so that
T is perpendicular to C.
For an example of its use see tools_input/gener_nanowire.in.
- gener_2d_slab.x reads a two dimensional
Bravais lattice index and atomic coordinates an generates an infinite ribbon
perpendicular to G = m
b1 + n
b2
, where
b1
and b2
are the primitive reciprocal lattice vectors
of the 2D Bravais lattice. The number of rows of the ribbon, and the number of
atoms per row are given as input variables.
For an example of its use see tools_input/gener_2d_slab.in.
- gener_3d_slab.x reads a three dimensional
Bravais lattice index and atomic coordinates and generates an infinite slab
perpendicular to G = m
b1 + n
b2 + o
b3
,
where b1
, b2
and b3
are the primitive reciprocal
lattice
vectors of the Bravais lattice. The number of layers of each slab, and the
size of the surface unit cell are given as input parameters.
For an example of its use see tools_input/gener_3d_slab.in.
- hex_trig.x reads the values of a
and c
of the conventional hexagonal cell of a rhombohedral lattice (in Ångstrom),
and gives as output the size ar
(in a.u.) and the cosine of the angle
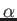 of the rhombohedral cell. This information can
be written in the input of pw.x for this type of cells. It
is used to convert the structural information contained in a CIF file
to the pw.x input.
of the rhombohedral cell. This information can
be written in the input of pw.x for this type of cells. It
is used to convert the structural information contained in a CIF file
to the pw.x input.
- kovalev.x writes the correspondence between point group
symmetry operations defined in the Kovalev tables and those used by QUANTUM ESPRESSO.
- mag_point_group.x gives a few information on the magnetic point
groups.
- optical.x contains a few utilities for
optical properties calculations. It transforms a complex dielectric
constant into a complex index of refraction and computes the reflectivity
or the absorption coefficient for cubic system. It converts also from
energy of the photon in eV to the frequency in Hz or the wavelength in nm.
- pdec.x reads the temperature dependent elastic constants files
calculated for several pressures and makes a plot of the elastic constants as
a function of pressure at several temperatures.
- plot_sur_states.x reads the dump file produced
by thermo_pw in a what='scf_2d_bands' calculation that
contains the planar averages of all the states, and plots
the states with the k point and the band numbers requested in input.
For an example of its use see tools_input/plot_sur_
states.in.
- rotate_tensors.x applies a rotation to a tensor
of rank 1, 2, 3, or 4 defined in a coordinate system 1 and finds the form
of the tensor in a new coordinate system 2.
- space_groups.x gives several information on space groups.
It can give the names of the space group given the number reported in
the International Tables for Crystallography (ITA), or
the number given one of the names, translate the names between different
editions of the ITA tables or the Shönflies name. It gives the list
of coset representatives of each space group and the list of symmorphic
space groups.
For an example of its use see tools_input/space_groups.in.
- supercell.x reads a three dimensional
Bravais lattice index and the atomic coordinates of the atoms inside a unit
cell and produces a supercell with
n1 x n2 x n3
cells of
the original
unit cell or a supercell delimited by three arbitrary Bravais lattice
vectors given in crystal or cartesian coordinates. For centered cells
there is the option to consider n1
, n2
,
and n3
for the primitive or for the centered Bravais lattices.
The input unit cell can be specified also by giving the space group and
the coordinates of the nonequivalents atoms.
It can be useful to study defects or to calculate all the
atomic positions starting from the space-group and the
nonequivalent positions.
It is also possible to give the input Bravais lattice by using
ibrav=0 and the three principal vectors. In this case,
before generating the supercell, the code rotates the Bravais
lattice so that it has the same orientation of the vectors described
in the thermo.pdf guide.
For an example of its use see tools_input/supercell.in.
- test_colors.x produces a postscript file with the
gnuplot colors that can be used in the plots.
- translate.x reads a set of atomic positions and a translation
vector and translates the atomic positions. It can read also a rotation matrix
and roto-translate the atomic positions.
- units.x writes on output the numerical constants used
to write the guide units.pdf and equilibrium.pdf.
It computes also the error associated to each conversion factor.
For a detailed description of the input variables please look at the beginning
of the fortran sources of each code.
Next: 7 Examples, examples_qe, inputs,
Up: User's Guide for the
Previous: 5 Restarting an interrupted
Contents
2021-07-20