Fahimeh Baftizadeh
 I got my B.Sc. (Physics) and M.Sc. (Condese Matter Physics) at Ferdowsi University of Mashhad in Iran. I am very much interested in investigating the physics of the living systems and that is why I am now in the third year of my PhD in "Statistical and biological physics" at SISSA in Trieste, Italy.
My PhD project is focused on "MD Simulations of polypeptide aggregation". This choice was inspired by the link that exist between the propensity of proteins to aggregate with the onset of several neurodegenerative disorders such as Alzheimer, Hungtinton and Parkinson. Understanding the structure of the aggregates and the thermodynamic details of the aggregation processes would be of great importance for the rational design of drugs against these diseases. At the present time, these issues still remain open and unsolved!
I got my B.Sc. (Physics) and M.Sc. (Condese Matter Physics) at Ferdowsi University of Mashhad in Iran. I am very much interested in investigating the physics of the living systems and that is why I am now in the third year of my PhD in "Statistical and biological physics" at SISSA in Trieste, Italy.
My PhD project is focused on "MD Simulations of polypeptide aggregation". This choice was inspired by the link that exist between the propensity of proteins to aggregate with the onset of several neurodegenerative disorders such as Alzheimer, Hungtinton and Parkinson. Understanding the structure of the aggregates and the thermodynamic details of the aggregation processes would be of great importance for the rational design of drugs against these diseases. At the present time, these issues still remain open and unsolved!
Research project: atomistic simulation of aggregation of small polipeptides
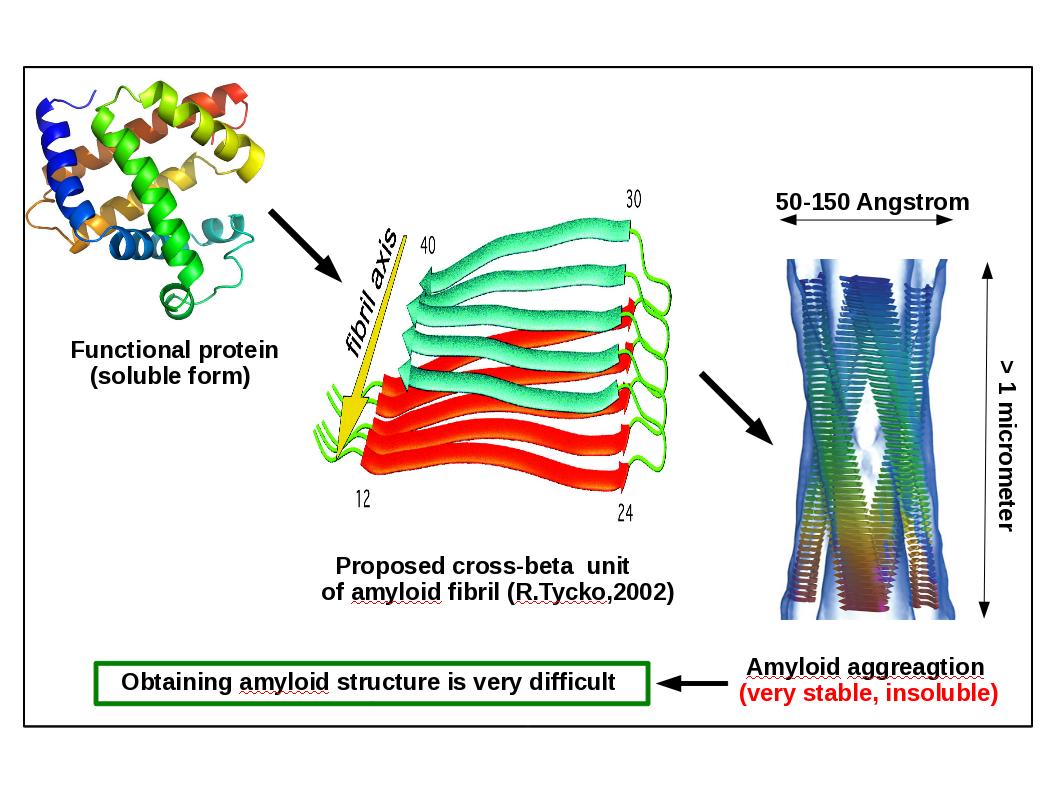 Under certain conditions proteins can convert from their soluble and biologically active forms into highly orderedand insoluble fibrillar aggregates with a high content of beta sheets. This can give rise to severe pathologies ranging from neurodegenerative disorders to systematicamyloidoses . There are more than 20 diseases related to amyloid fibrils such as Alzheimer, Parkinson, Huntington and Prion (mad cow) [Chiti06,Fandrich02].
Amyloid fibrils are non crystalline and insoluble and thus are not amenable to analysis by conventional X-ray crystallography and solution NMR. Therefore, a detailed description of amyloid formation at the molecular level remains elusive [Chiti06,Fandrich02], and computer simulations are a natural tool to approach this problem.
Due to the complexity of the system so far mainly intermediate-resolution models with implicit solvent or coarse grained model have been used to study aggregation [Auer07,Cheon07, Wang08].
Under certain conditions proteins can convert from their soluble and biologically active forms into highly orderedand insoluble fibrillar aggregates with a high content of beta sheets. This can give rise to severe pathologies ranging from neurodegenerative disorders to systematicamyloidoses . There are more than 20 diseases related to amyloid fibrils such as Alzheimer, Parkinson, Huntington and Prion (mad cow) [Chiti06,Fandrich02].
Amyloid fibrils are non crystalline and insoluble and thus are not amenable to analysis by conventional X-ray crystallography and solution NMR. Therefore, a detailed description of amyloid formation at the molecular level remains elusive [Chiti06,Fandrich02], and computer simulations are a natural tool to approach this problem.
Due to the complexity of the system so far mainly intermediate-resolution models with implicit solvent or coarse grained model have been used to study aggregation [Auer07,Cheon07, Wang08].
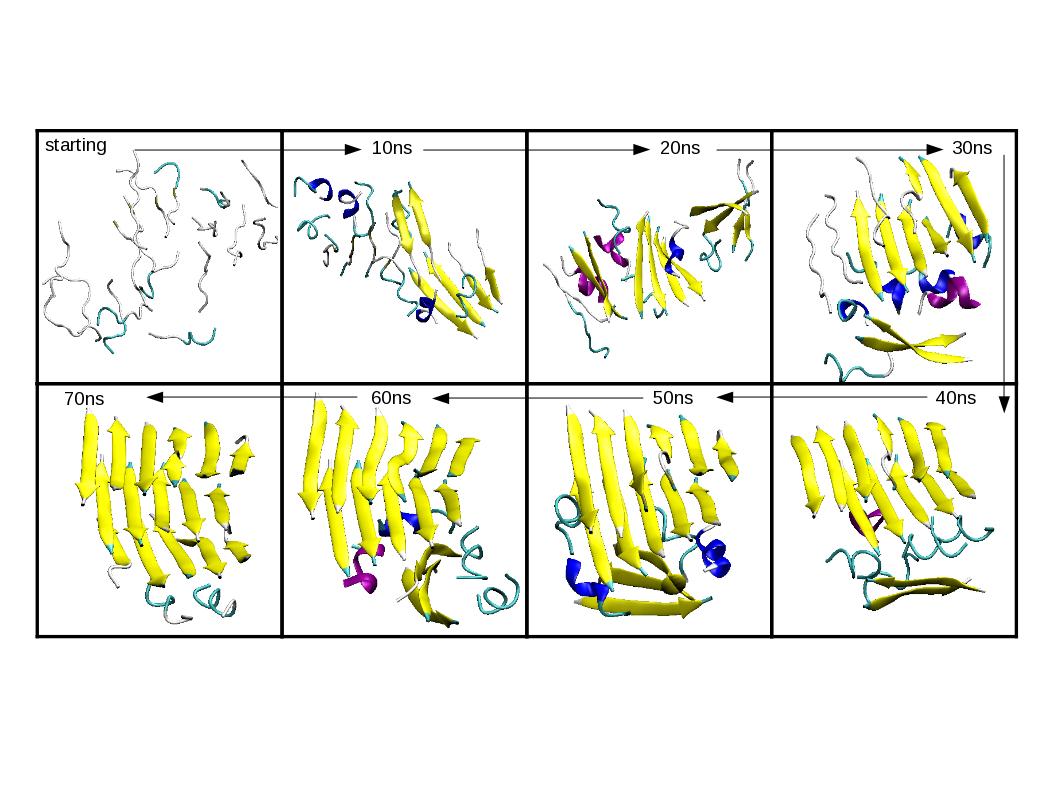 To get some insights into this process we performed accurate atomistic simulations, with an all-atom force-field describing explicitly both the polypeptides and the solvent and using bias-exchange metadynamics, which has already been successfully used to study protein folding and ligand docking. We simulate a system of 18 chains of 8-Valine in explicit solvent. This choice was guided by the "general hypothesis" of amyloid formation, according to which the ability to assemble into ordered cross-beta structure is an inherent characteristic of polypeptide chains that does not depend on their sequence. With our algorithm we obtained a large number of aggregated structures with a significant content of beta sheets (see figure). We were able to measure relevant observables including entropy and enthalpy. Preliminary results indicate the presence of a large enthalpic barrier separating the disordered aggregate from the ordered cross-beta structure.
To get some insights into this process we performed accurate atomistic simulations, with an all-atom force-field describing explicitly both the polypeptides and the solvent and using bias-exchange metadynamics, which has already been successfully used to study protein folding and ligand docking. We simulate a system of 18 chains of 8-Valine in explicit solvent. This choice was guided by the "general hypothesis" of amyloid formation, according to which the ability to assemble into ordered cross-beta structure is an inherent characteristic of polypeptide chains that does not depend on their sequence. With our algorithm we obtained a large number of aggregated structures with a significant content of beta sheets (see figure). We were able to measure relevant observables including entropy and enthalpy. Preliminary results indicate the presence of a large enthalpic barrier separating the disordered aggregate from the ordered cross-beta structure.
[Chiti06] F. Chiti, C.M. Dobson, Annu. Rev. Biochem 75 (2006) 333-366
[Fandrich02] M. Fandrich, C. M. Dobson, EMBO J., 21 (2002) 5682--5690
[Fandrich02] M. Fandrich, C. M. Dobson, EMBO J., 21 (2002) 5682--5690
[Auer07] S. Auer, C. M. Dobson, M. Vendruscolo, HSFP J., 1 (2007) 137-146
[Cheon07] M. Cheon et al, PLOS Computational Biology 3 (2007) 1727-1738
[Wang08] J. Wang et al, Biophysical journal 95 (2008) 5037-5047