Pilar Cossio
 I am a former Physics student at the Univeristy of Antioquia, Colombia. Currently, I am working in as a PhD student in the Statistical and Biological Physics Sector at the International School for Advanced Studies (SISSA), in Trieste, Italy. I work mainly in developing methodologies for computational studies of protein folding and protein design.
I am a former Physics student at the Univeristy of Antioquia, Colombia. Currently, I am working in as a PhD student in the Statistical and Biological Physics Sector at the International School for Advanced Studies (SISSA), in Trieste, Italy. I work mainly in developing methodologies for computational studies of protein folding and protein design.
Research topics:
Exploration of protein conformational space:
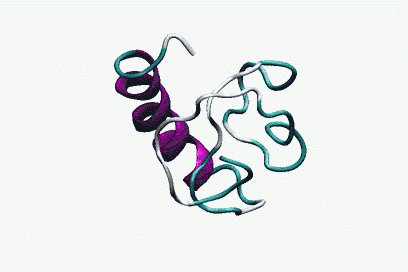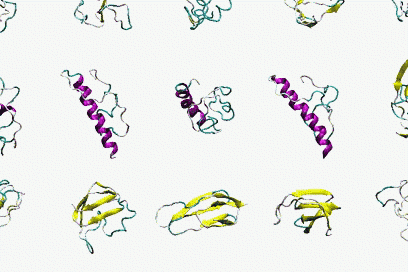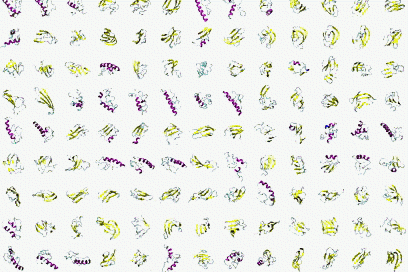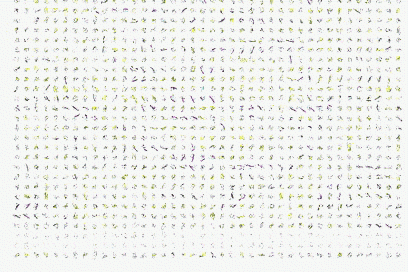 |
In collaboration with Antonio Trovato, Flavio Seno and Amos Maritan, Padova, by using Bias Exchange Metadynamics we performed an exhaustive exploration of the conformational space of a 60 amino acid polypeptide chain described with an accurate all-atom interaction potential. We generated a database of around 30,000 compact folds with at least 30% of secondary structure corresponding to local minima of the potential energy. This ensemble plausibly represents the universe of protein folds of similar length: indeed, all the known folds are represented in the set with good accuracy. However, we discover that the known folds form a rather small subset, which cannot be reproduced by choosing random structures in the database. Rather, natural and possible folds differ by the contact order, on average significantly smaller in the former. This suggests the presence of an evolutionary bias, possibly related to kinetic accessibility, towards structures with shorter loops between contacting residues.
P Cossio, A Trovato, F Pietrucci, F Seno, A Maritan, A Laio,
Exploring the Universe of Protein Structures beyond the Protein Data Bank
PLOS COMPUTATIONAL BIOLOGY, 6, e1000957 (2010)
Folding of small peptides:
In collaboration with Giulia Rossetti and Prof. Paolo Carloni, Julich, we have used Bias Exchange Metadynamics to characterized the thermodynamic and kinetic folding picture of a small N terminal- 17 a.a. fragment (Sequence: MATLEKLMKAFESLKSF). This peptide is involved in Huntington disease and has been showed to produce an increase of polyQ fibrillation.
Quality assessment of the different similarity measures for protein structures:
In collaboration with Fabio Pietrucci, CECAM Lausanne, we analyzed microseconds-long all-atom molecular dynamics simulations of a polypeptide, in order to characterize the quality of different similarity measures for protein structures. We find that a distance based on backbone dihedral angles performs very well in distinguishing structures that are kinetically correlated from those that are not, while the widely used Ca root mean square distance performs more poorly. The root mean square difference between contact matrices turns out instead to be the metric providing the highest clustering coefficient, namely, according to this similarity measure, the neighbors of a structure are also, on average, neighbors among themselves. From our findings we have proposed a distance measure which is optimal both for distinguishing structures which are distant in time and for giving a consistent cluster analysis.
Protein Design:
In collaboration with Flavio Seno and Antonio Trovato, Padova, we are designing semi-empirical potentials in order to discriminate a native folded state from a decoy set. These potentials are learned from the Protein Data Bank with statistical physics techniques. After optimizing these potentials, the goal is to use them together with simulational techniques (like Monte Carlo) to design a new protein fold.