Il programma hydrogen_gauss risolve il problema secolare per l'atomo di idrogeno utilizzando due diverse basi non ortonormali:
| (5.91) |
| (5.92) | |||
| (5.93) | |||
| (5.94) |
| (5.95) |
Il programma permette di assegnare gli esponenti ![]() ,
sotto forma di larghezze
,
sotto forma di larghezze ![]() delle gaussiane:
delle gaussiane:
| (5.96) |
Il codice procede quindi a valutare tutti gli elementi
delle matrici ![]() e
e ![]() . Il calcolo è basato
sulle espressioni degli integrali calcolati analiticamente.
In particolare si trova per l'onda S
. Il calcolo è basato
sulle espressioni degli integrali calcolati analiticamente.
In particolare si trova per l'onda S
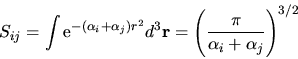 |
(5.97) |
![\begin{displaymath}
H^K_{ij} = \int {\rm e}^{-\alpha_i r^2} [-\nabla^2]
{\rm e}^...
..._i+\alpha_j}
\left(\frac{\pi}{\alpha_i+\alpha_j}\right)^{3/2}
\end{displaymath}](img846.gif) |
(5.98) |
| (5.99) |
Il codice procede dunque a chiamare la subroutine diaghg che risolve il problema secolare generalizzato (ossia applica il principio variazionale), ritornando il vettore e contenente gli autovalori in ordine crescente di energia, e la matrice v contenente gli autovettori, ossia i coefficienti dell'espansione.
Al suo interno, diaghg effettua il calcolo in due stadi descritto nella sezione precedente, delegando la risoluzione del problema agli autovalori semplice alla subroutine dsyev della libreria di algebra lineare LAPACK, ed utilizzando alcune chiamate addizionali a subroutines della libreria BLAS. Informazioni addizionali su queste librerie sono disponibili a partire dalla pagina del corso lapack.html.
Da notare che dopo la prima diagonalizzazione (relativa
a ![]() ) diaghg getta via tutti gli autovettori corrispondenti
ad autovalori nulli (entro una soglia di tolleranza numerica).
Questi corrispondono a combinazioni linearmente dipendenti dalle
altre. Il numero di vettori di base linearmente indipendenti
trovati viene riportato nell'output.
) diaghg getta via tutti gli autovettori corrispondenti
ad autovalori nulli (entro una soglia di tolleranza numerica).
Questi corrispondono a combinazioni linearmente dipendenti dalle
altre. Il numero di vettori di base linearmente indipendenti
trovati viene riportato nell'output.
Il programma procede quindi a scrivere i coefficienti
dell'espansione nel file s-coeff.out (p-coeff.out):
per ogni funzione ![]() , e la funzione d'onda dello stato
fondamentale nel file s-wfc.out (p-wfc.out).
, e la funzione d'onda dello stato
fondamentale nel file s-wfc.out (p-wfc.out).