Ligand-protein docking by accurate atomistic simulations
Enzymes
are protein molecules which catalyze important chemical reactions in
our body. To perform their action, enzymes bind to other molecules
called ligands or substrates. Sometimes, enzymes must be blocked to
prevent them to cause diseases, like in the case of HIV-1 protease
which is involved in AIDS. A small ligand (drug) is therefore designed
which binds to the enzyme blocking it. Often the mechanism by which the
drug binds to the enzyme is not known.
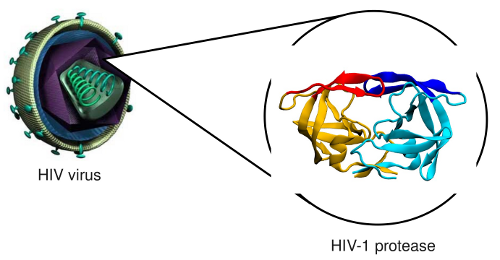
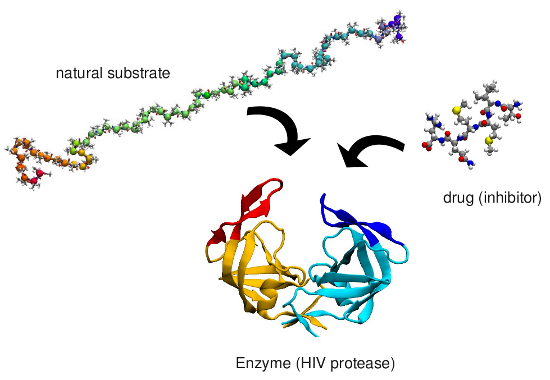
Binding mechanism of HIV-1 protease
We investigated the mechanism by which a small peptide substrate binds to
HIV-1 protease. To this aim, we performed long molecular dynamics
simulations (1.6 microsec) using an accurate explicit solvent force
field, and accelerating the exploration of 7 reaction coordinates by bias-exchange metadynamics.
We observed several times the ligand approaching the enzyme and binding
to it. The structure of the complex is in excellent agreement with the
crystallographic evidences. From the simulation we constructed a kinetic model
describing the stability of the intermediate states and the rates of
interconversion among them. The computed binding free energy and
association/dissociation rates are in agreement with available
experiments.
It turns out that opening of the protease flaps is not required for the
binding process, and that expulsion of the water molecules from the
enzyme cavity is a key kinetic step. We hope that the insight we
obtained on the binding pathway will help the rational design of more
effective drugs.
Our present research effort is devoted to make
this type of binding calculations easy to use and accessible to all
users of molecular dynamics programs.
Bibliography
Substrate Binding Mechanism of HIV-1 Protease from Explicit-Solvent Atomistic Simulations
JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, 131, 11811 (2009)
A Kinetic Model of Trp-Cage Folding from Multiple Biased Molecular Dynamics Simulations
PLOS COMPUTATIONAL BIOLOGY, 5, e1000452 (2009)
This page has been written by Fabio Pietrucci. Please also refer to his home page for further details.