Assessing the quality of folding and docking poses
The ability of discriminating the native pose of a protein among a large set of
incorrect ones (decoys) is a fundamental step in any protein structure prediction
process. This is usually accomplished via a scoring function, which associates to
every pose a free energy value (score) whose minimum should always correspond, in principle, to
the native pose. Since the physical laws underlying protein folding and protein-protein
interaction phenomena are the same, we believe a single method for treating both problems
could be devised.
BACH: Bayesian Analysis Conformation Hunt
BACH is a statistical knowledge-based potential trained on a set of ~450 folded globular monomers.
It makes use of a pairwise, residue-based energy term, an atomwise clash term and a solvation
contribution. The pairwise term has been recently refined including a difference between polar and
apolar contacts (BACH-SixthSense). Unlike the best performing state-of-the-art scoring functions,
BACH-SixthSense has proven able to reach top performances in both protein folding (CASP, fisa, 4state, ...)
and protein-protein interaction (CAPRI, ZDOCK-generated decoy sets) tests.
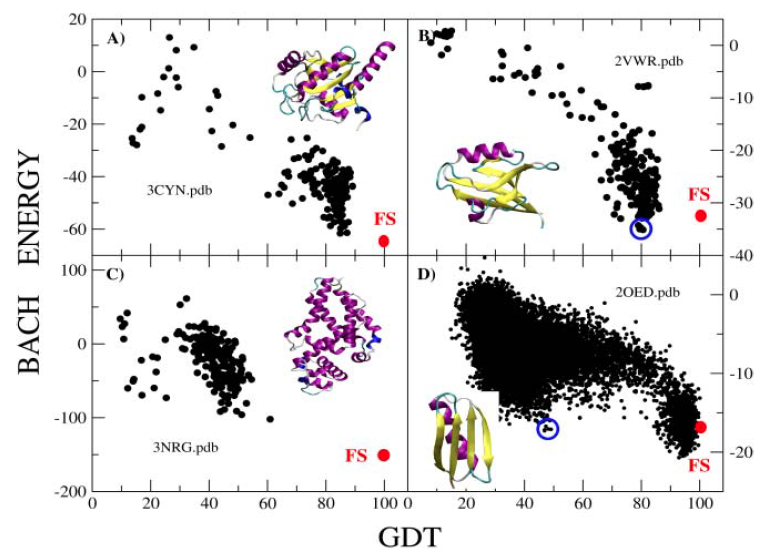
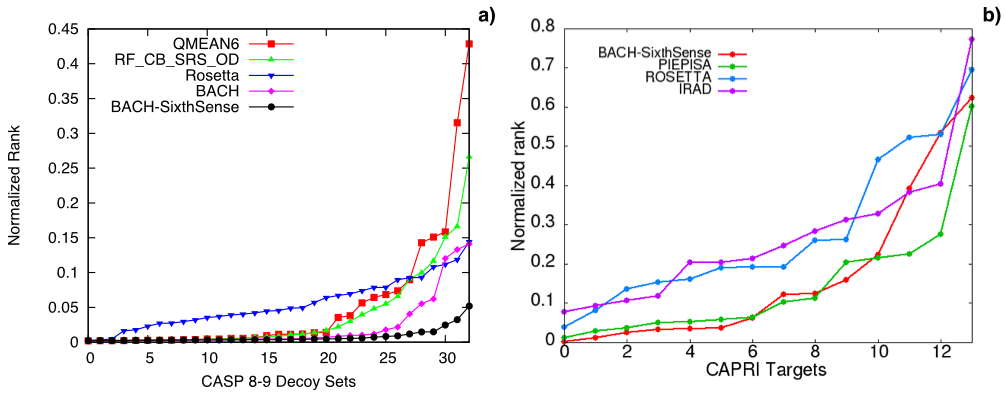
Here we report a comparison of the performance of BACH on 33 CASP and 14 CAPRI decoy sets. For each decoy set
we sort the scoring functions energy values from the lowest to the highest and see at which point of the rank
the native pose is found. The lower the rank, the better the performance.
Recently, BACH has also been adapted for working as a collective variable in enhanced sampling
methods (cfr. metadynamics).
Bibliography
A Simple and Efficient Statistical Potential for Scoring Ensembles of Protein Structures
SCIENTIFIC REPORTS, 2, 351 (2012)
BACHSCORE: a Tool for Evaluating Efficiently and Reliably the Quality of Large Sets of Protein Structures
COMPUTER PHYSICAL COMMUNICATIONS, 184, 2860 (2013)
Native Fold and Docking Pose Discrimination by a Single Residue-Based Scoring Function
(forthcoming)