Machine Learning
Automated training of neural-network interatomic potentials
We build automated tools that turn a handful of atomistic structures into reliable, production-ready neural-network interatomic potentials with minimal manual tuning. AiiDA-TrainsPot Digital Discovery (2026) orchestrates DFT calculations, systematic data augmentation, active-learning MD, and a calibrated committee-disagreement uncertainty estimator so we can safely identify which new configurations require expensive ab-initio labels. The modular platform supports training from scratch or fine-tuning foundation models with different training engines, preserves full data provenance, and has been validated on dynamics, defects, amorphous phases, and composition-driven phase transitions—delivering near-DFT accuracy at a fraction of the cost.
Spectral operator representations
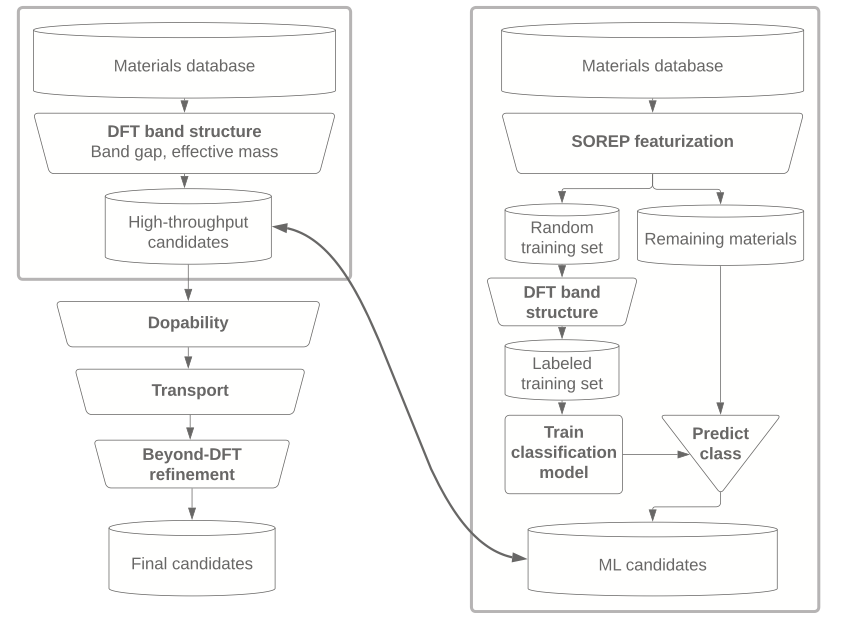
We develop physics-informed machine-learning representations that describe materials through their electronic structure rather than atomic geometry alone. In npj Computational Materials 10, 278 (2024), we introduce Spectral Operator Representations (SOREPs), a general framework that transforms approximate electronic-structure models into compact, symmetry-preserving, and interpretable descriptors. By projecting physical operators onto model electronic states and learning from their spectra, SOREPs naturally capture non-local and intrinsically electronic properties such as band gaps and carrier mobilities. We demonstrate their effectiveness for materials similarity, polymorph discrimination, and accelerated materials screening. Applied to transparent-conducting materials, SOREPs enable machine-learning models that recover most promising candidates while requiring DFT calculations for only a small fraction of a large database. This work reflects ARGO's broader effort to combine electronic-structure theory, machine learning, and automation to enable scalable and physically grounded materials discovery.