Atomistic simulations are becoming increasingly useful, as they
have the potential to investigate physical processes with a
resolution which cannot be achieved by experiments.
Still, interesting events such as chemical reactions, protein folding, phase transitions, etc., happen on a time scale that is enormously long for computer simulations.
Several methods
have been developed to cope with this problem, for example
thermodynamic integration, free energy perturbation,
parallel tempering, Jarzynski's identity-based methods,
steered MD, etc.
 |
In our group we work at improving and extending these techniques in order to make them suitable for studyng realistic processes. In protein folding, protein aggrefgation and protein-protein interaction the number of "interesting" variables that one has to sample and explore is intrinsically very large. This has led to develop a new method, bias-exchange metadynamics, that allows the simultaneous reconstruction of a free energy in several variables.
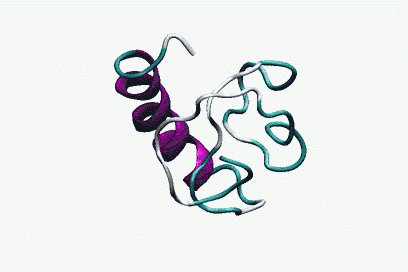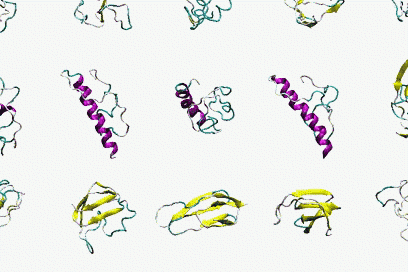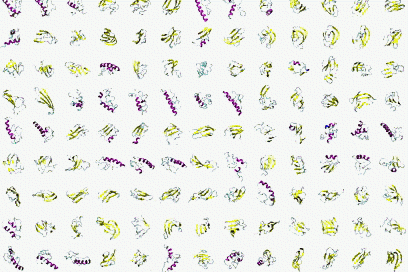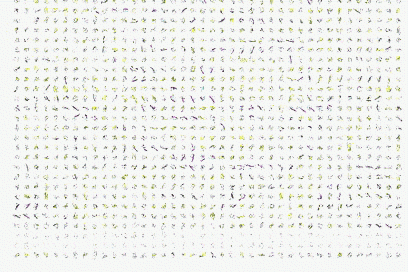
This approach allows predicting the folded state and the folding time of small proteins (up to 60 amino acids) described with an accurate potential, in which the water is described explicitly (see Foding for more details).
The same approach can be used for studying, also with a very accurate potential, the binding process of drugs to their target protein, predicting with great accuracy the binding affinity (see Docking for more details).
We are also working on the development of an approach aimed at designing peptides capable of binding selectively and strongly to a target molecule, for example a drug or another protein, whose concentration has to be detected. In order to design the peptides reliably and efficiently we are developing a novel scoring function, capable of discriminating the correct structure of a protein, and of a protein-protein complex (see Scoring protein structures> for details).